Past Research
TAP: An Evaluation Measure for Genetic Sequence Database Retrieval (Carroll et al., 2010a, 2010b)

|
Retrieval is a common task in many different areas (e.g., web searches, genetic sequence similarity searches, and biomedical abstract text searches). In many fields, a threshold is often applied to the retrieved results. Studies using genetic sequence similarity searches often use pooled receiver operating characteristic (ROC) curves to evaluate the performance of different algorithms. Unfortunately, this method does not accurately reflect the actual usage of the retrieval algorithms. Furthermore, it is susceptible to being arbitrarily skewed by a single query. To remedy these issues, we introduced the Threshold Average Precision (TAP), a method based on average precision (from information retrieval) that explicitly incorporates E-values. Along with this method, we proposed additional criteria for ideal retrieval methods. The TAP measure is a robust method that addresses these criteria. Our TAP web server is available at http://www.ncbi.nlm.nih.gov/CBBresearch/Spouge/html.ncbi/tap/tap.cgi. This criterion has been well received by researchers. For example, it was adopted as the single evaluation measure for a task at the BioCreAtIvE III workshop.
I lead this project with support from others at the National Institutes of Health (NIH).
Identification of Greek Papyri Fragments using Genetic Sequence Alignment Algorithms (Williams et al., 2014a; see also Williams et al., 2014b, 2015)


|
Papyrologists analyze, transcribe, and interpret damaged papyri fragments in order to better understand the linguistics, culture, and literature of the ancient world. One of their common tasks is to determine if the fragment matches a known manuscript. This is especially challenging when the fragment contains only limited information (e.g., due to deterioration). In the last 100 years, only about 2% of the 500,000 fragments recovered from the Egyptian city Oxyrhynchus have been edited and published and only about 10% have even been examined by professionals and given preliminary identifications. We do not know what new ancient texts might be found once these documents are curated and published, but using current methods this process will take in excess of 1000 years.
In bioinformatics, the process of finding relationships among genes is confronted with a similar problem to that of determining the identity of papyri fragments. Genes are often represented by a sequence of continuous letters (comparable to the format of Greek papyri fragments). Relationships are inferred by finding multi-letter patterns shared between two genetic sequences. This process is commonly referred to as genetic sequence alignment. In order to enable papyrologists with the ability to transcribe severely damaged texts, we investigated the applicability of genetic sequence alignment algorithms as a method for identifying Greek papyri fragments. Sequence alignment algorithms leverage a new form of non-contextual, multi-line text identification for the Greek language that can greatly accelerate the tedious task of transcription. Our implementation of these methods is able to process the text of thousands of fragments per second and has been adopted by papyrologists at the University of Oxford.
This project was funded by a MTSU FRCAC grant, of which I was the PI. I mentored a Masters student, Alex Williams, in conjunction with Dr. John Wallin in the Physics and Astronomy Department at MTSU.
False Discovery Rates for Genetic Sequence Searches (Carroll et al., 2013, 2015)
|
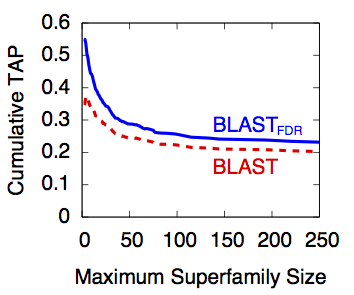
Over the past few decades, discovery based solely on genetic sequence similarity has become a widely accepted practice. Consequently, comparative accuracy in genetic sequence alignment algorithms (GSAAs) has been rigorously studied for improvement. Unlike most components of these algorithms, the E-value threshold criterion has yet to be thoroughly investigated at the same depth. An investigation of the threshold is necessary as it exclusively dictates which sequences are deemed statistically relevant and irrelevant.
We replaced the uniform threshold criterion commonly found in genetic sequence search algorithms with the false discovery rate (FDR) statistic in order to strengthen their efficacy. Using the BLAST and PSI-BLAST software packages (Camacho et al., 2009), we demonstrated the applicability of such a replacement in both non-iterative (BLASTFDR) and iterative (PSI-BLASTFDR) searches. Using the Threshold Average Precision, BLASTFDR yielded 14.1% better retrieval performance than BLAST on a large (5,161 queries) test database and PSI-BLASTFDR attained 11.8% better retrieval performance than PSI-BLAST.
I mentored three undergraduate students for this project and worked with Dr. John Spouge at the NIH.
Interactive Toxicology Database

|
Currently, information about the toxicity of each chemical and how it reacts with other compounds is housed around the world in databases held by government organizations and privately within corporations. We formed agreements with the organizations of several of the largest toxicology databases to bring their data together into a single database. This will allow researchers to easily obtain more information about the chemicals that they are studying for regulatory and production purposes. We facilitated this by designing a flexible semantic MySQL database and making it available through a web-interface. Furthermore, we developed the functionality for users to filter the database for downstream analysis of those results.
This project was funded by the Health and Environmental Sciences Institute. I worked in collaboration with Dr. Ryan Otter from the Department of Biology at MTSU and Dr. Joshua Phillips from the Department of Computer Science at MTSU. Additionally, Dr. Phillips and I co-mentored an undergraduate and a graduate student on this project.
Gene Family Alignment Visualization ServerGenes typically consist of dozens of exons, which are the portion of the genetic code that is eventually translated into proteins. Exon sequence size and nucleotide identity are under selective constraints because they must maintain this coding potential for proteins. Therefore, exon similarity can be used to examine relatedness of gene families that have arisen by gene or genome duplication. Currently, researchers are required to manually align the exon portions of gene families to visualize and understand their relationships. The process is usually avoided due to its tedious and time consuming nature. There has been an expressed need of a visualization of the alignment of gene families to assist in unraveling biological complexities. This visualization server automatically aligns the exons in each DNA sequence specified by the user and displays those results. This visualization allows the human mind to quickly make inferences and detect patterns from the hundreds of data points that are hidden due to complexity in the system.
For this project, I collaborated with Dr. Rebecca Seipelt-Thiemann from the Department of Biology at MTSU and mentored some undergraduate students, including two that were funded by Undergraduate Research Experience and Creative Activity awards from MTSU.


|
Benchmark evaluations are necessary to test the relative performance of an algorithm to variants and competing algorithms. Benchmarking is commonly performed for genetic sequences with just a single domain (the basic unit of a protein). While existing single domain databases provide an invaluable foundation for evaluation, they fail to capture the true biological complexity inherent in genetic sequences. We have developed a database suite of multiple domain sequences and the scripts to evaluate their performance in a single machine or cluster environment. Additionally, we provide a case study, using the database to evaluate the performance of three alignment algorithms. This database will be helpful to accurately evaluate future genetic sequence alignment algorithms.
I lead this project and collaborated with Drs. John Spouge and Mileidy Gonzalez at the National Center for Biotechnology Information.
Iterative Genetic Sequence Alignment Using Multiple Blocks
|
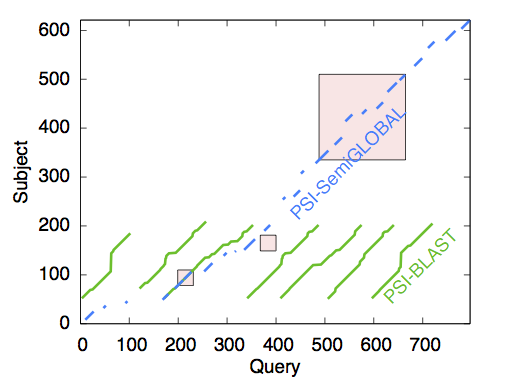
Every second of every day, researchers use genetic sequence alignment algorithms (GSAAs) to find sequences that are biologically similar to a query sequence. They utilize these results for genome annotation, 2D and 3D structure prediction, and to determine biological relationships between organisms among other uses. Most GSAAs find "local" alignments (maximal similarity between substrings). On the other hand, using "semi-global" alignment can provide more accurate statistical estimators because it takes advantage of the inherent domain structures in sequences. We developed the PSI-SemiGLOBAL algorithm that uses multiple blocks in the query sequences to look for biologically related sequences in a large database. Results indicate that PSI-SemiGLOBAL's statistical score is more accurate than the commonly used "local" search algorithms (e.g., RPS-BLAST (Schaffer et al., 1999) and HMMER (http://hmmer.janelia.org)). To make execution time for PSI-SemiGLOBAL practical, we leveraged the search and database heuristics from the BLAST framework (Camacho et al., 2009).
I lead this project and collaborated with Drs. John Spouge and Mileidy Gonzalez at the National Center for Biotechnology Information.
Discoveries about the Karenia Brevis Chloroplast Genome (Klinger
|
Large concentrations of K. brevis are responsible for algal blooms in the Gulf of Mexico. Understanding their chloroplast genome and transcriptome is important because these blooms produce significant levels of toxins that are harmful to ocean flora and fauna and to humans. The chloroplast genome of several closely related species are comprised of multiple 2,000 to 10,000 residue "mini-circles" with one to four genes while the structure of K. brevis' chloroplast is unknown. We have assembled RNA-Seq reads of K. brevis and are exploring the possibility that they are transcribed from mini-circles. We have already found evidence for several genes that were not previously known to be in the chloroplast genome. Furthermore, we have data that supports a novel RNA post-transcription modification of poly-A 5' additions paired with poly-U tails.
For this project, I mentored three PhD students in the Computational Science (COMS) program at MTSU and worked in conjunction with Dr. A. Bruce Cahoon from the Department of Natural Sciences at The University of Virginia's College at Wise.
References
- C. Camacho, G. Coulouris, V. Avagyan, N. Ma, J. Papadopoulos, K. Bealer, and T.L. Madden. 2009. BLAST+: architecture and applications. BMC bioinformatics 10:421.
- H.D. Carroll, M.G. Kann, S.L. Sheetlin, and J.L. Spouge. 2010. Threshold Average Precision (TAP-k): A Measure of Retrieval Efficacy Designed for Bioinformatics. Bioinformatics 26:14, 1708-1713. (web, pdf)
- H.D. Carroll, M.G. Kann, S.L. Sheetlin, and J.L. Spouge. 2010. Threshold Average Precision (TAP-k): A Retrieval Efficacy Measure for Bioinformatics. Intelligent Systems for Molecular Biology. (web, pdf)
- H.D. Carroll, J.L. Spouge, and M.W. Gonzalez. 2019. The MultiDomainBenchmark Suite: a multiple-domain query and subject database suite. BMC Bioinformatics 20:77. (web)
- H.D. Carroll, A.C. Williams, A.G. Davis, and J.L. Spouge. 2013. False Discovery Rate for Homology Searches. In Advances in Bioinformatics and Computational Biology, J. C. Setubal, and N. F. Almeida (Eds.) 8213, 194-201. (web, pdf)
- H.D. Carroll, A.C. Williams, A.G. Davis, and J.L. Spouge. 2015. Improving Retrieval Efficacy of Homology Searches using the False Discovery Rate. IEEE Transactions on Computational Biology and Bioinformatics 12, 531-537. (web, pdf)
- C.M. Klinger, L. Paoli, R.J. Newby, M.Y. Wang, H.D. Carroll, J.D. Leblond, C.J. Howe, J.B. Dacks, C. Bowler, A.B. Cahoon, R.G. Dorrell, and E. Richardson. 2018. Plastid Transcript Editing across Dinoflagellate Lineages Shows Lineage-Specific Application but Conserved Trends. Genome Biology and Evolution 10:4, 1019-1038. (web)
- A.A. Schäffer, Y.I. Wolf, C.P. Ponting, E.V. Koonin, L. Aravind, and S.F. Altschul. 1999. IMPALA: matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics 15:12, 1000-1011.
- A.C. Williams, H.D. Carroll, J.F. Wallin, J. Brusuelas, L. Fortson, A. Lamblin, and H. Yu. 2014. Identification of Ancient Greek Papyrus Fragments Using Genetic Sequence Alignment Algorithms. In 10th IEEE International Conference on e-Science 2, 5-10. (web, pdf)
- A.C. Williams, J.F. Wallin, H. Yu, M. Perale, H.D. Carroll, A. Lamblin, L. Fortson, D. Obbink, C.J. Lintott, and J.H. Brusuelas. 2014. A Computational Pipeline for Crowdsourced Transcriptions of Ancient Greek Papyrus Fragments. In IEEE International Conference on Big Data, 100-105. (web, pdf)
- A.C. Williams, A. Santarsiero, C. Meccariello, G. Verhasselt, H.D. Carroll, J.F. Wallin, D. Obbink, and J. Brusuelas. 2015. Proteus: A Platform for Born Digital Critical Editions of Literary and Subliterary Papyri. In 2015 Digital Heritage 2, 453-456. (web, pdf)